Projects

Deidentification Pipeline (M2P2)
Multilingual de-identification techniques for medical records.

Liquid Biopsy
Computational solutions to improve liquid biopsies

Single-cell multi-omics analysis
Investigating skin cancers using single-cell multi-omics

AttentionDDI
Predicting drug drug interactions for enhancing patient safety

BEDICT models
Predicting base editing efficiencies in both lab cells and living models

Drug Synergy Prediction
Predicting drug synergy for enhancing therapeutic effectiveness

Emergency care outcome prediction
ML models rapidly stratify patient cardiac risk in emergency settings

Metagenomic Mining
Training AI algorithms on metagenomic data for predicting CAS9 PAM motifs

Nursing Resource allocation
ML models for predicting patient-related nursing workload.

Prediction of lung function
Deep learning models quantify lung disease progression

PRIDICT
Predicting prime editing efficiencies and optimizing prime editing guide RNA (pegRNA) design

Systemic Sclerosis Subtype Analysis
Generative DL models patient trajectories

TEEP
TnpB editing efficiency predictor

Alignment in Spoken Interaction
Studying how conversational coordination shapes meaning in human and machine communication

Data-driven psychiatry
Modeling speech and behavior to support mental health research and assessment

Modeling Spoken Language Beyond Text
Audio-first approaches to understanding language directly from speech

Protein Design
Generative AI for protein fitness optimization

Automated Image Analysis
AI-assisted diagnosis in rheumatology

Federated Learning
Today, in every aspect of our lives, everything we do leaves a digital footprint. Health-care institutions are also increasingly …

Human Reproduction Reloaded
We are proud to be part of the UZH University Research Priority Programs (URPP) where we together with the Schwank lab investigate the …
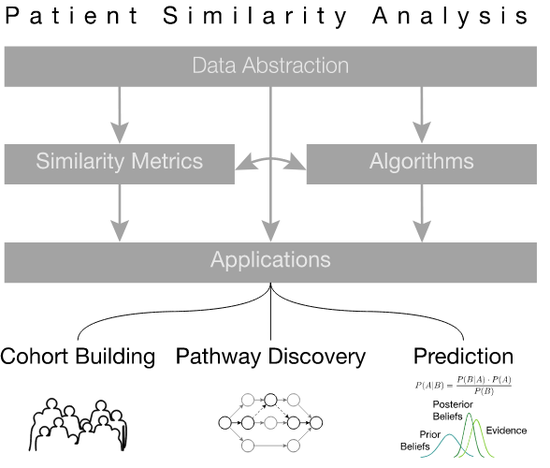
Patient Similarity Analysis
Since the early days of computing, healthcare professionals have dreamt of using the vast storage and processing powers of computers to …

Long-read sequencing - Part I
An introduction to long-read sequencing.
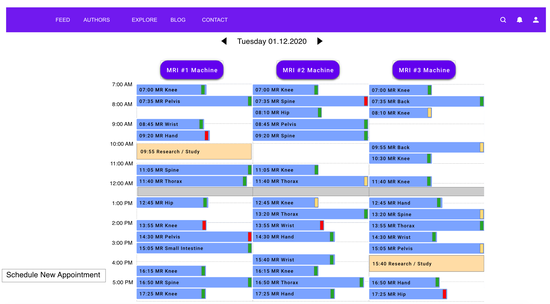
MRIdle
Helping reduce idle time in the USZ Radiology department

30 days All-Cause Readmission
Comparing neural-networks versus logistic regression for predicting readmission.
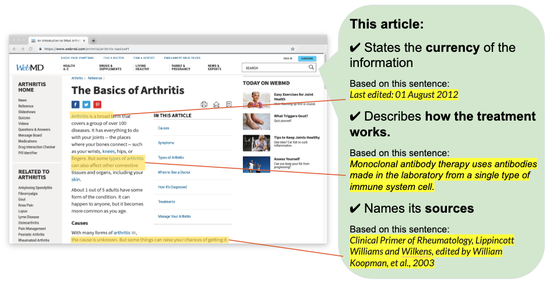
autoDISCERN
Assessing the quality of online health information with AI.
Single Cell RNA Sequencing
Cancer on the cell level.

Drug-Drug Interactions
Novel computational method for drug-drug interaction predictions which are an important consideration for patient treatment.
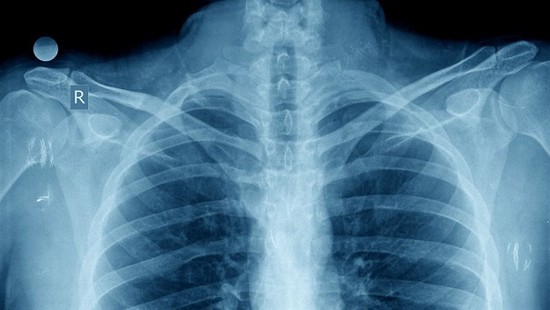
Automated Reports from Xrays
Using deep learning for automatically generated medical reports describing radiological images.